
Polarons from First Principles, without Supercells
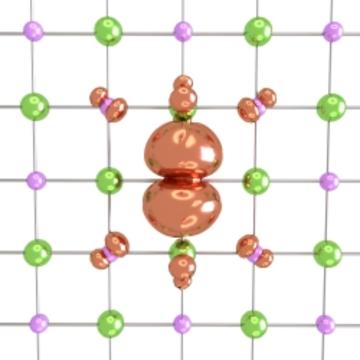
Weng Hong Sio and colleagues from the Materials Modelling and Design Group led by Professor Giustino have recently published papers in Physical Review Letters and Physical Review B which report a new formalism for modelling polarons. The new method combines elements of idealized mathematical models and numerical methods based on density-functional theory. A Physics viewpoint highlights that by reducing the calculations to within a single primitive crystal cell this new method opens up exciting prospects for studying more complex materials beyond the reach of previous models.