
Accurate and efficient computation of optical absorption spectra of molecular crystals
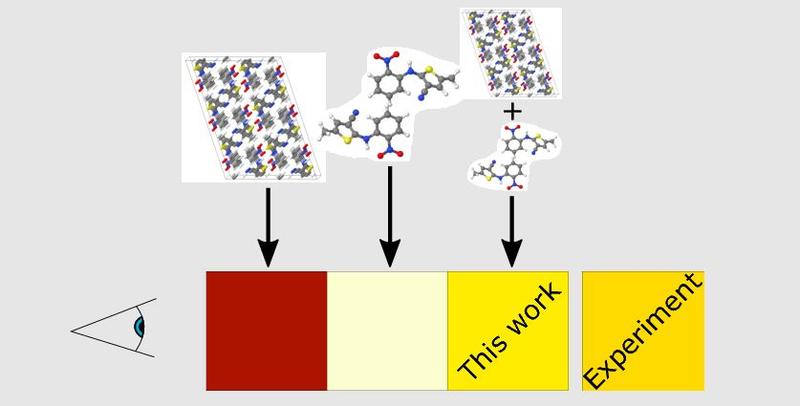
When calculating the optical absorption spectra of molecular crystals from first principles, the influence of the crystalline environment on the excitations is of significant importance. For such systems, however, methods to describe the excitations accurately can be computationally prohibitive due to the relatively large system sizes involved.
In this work, published in the Journal of Chemical Theory and Computation, Joseph Prentice (Materials, Oxford) and Arash Mostofi (Materials and Physics, Imperial College London) demonstrate a method that allows optical absorption spectra to be computed both efficiently and at high accuracy. This involves calculating the absorption spectrum of a supercell of the full molecular crystal using semi-local time-dependent density functional theory (TDDFT), before warping the spectrum using a transformation derived from smaller-scale semi-local and hybrid TDDFT calculations on isolated dimers.
The power of this method is demonstrated on three polymorphs of the well-known colour polymorphic compound ROY and findings that it outperforms both small-scale hybrid TDDFT dimer calculations and large-scale semi-local TDDFT supercell calculations, when compared to the experiment.